- Barajar
ActivarDesactivar
- Alphabetizar
ActivarDesactivar
- Frente Primero
ActivarDesactivar
- Ambos lados
ActivarDesactivar
- Leer
ActivarDesactivar
Leyendo...
Cómo estudiar sus tarjetas
Teclas de Derecha/Izquierda: Navegar entre tarjetas.tecla derechatecla izquierda
Teclas Arriba/Abajo: Colvea la carta entre frente y dorso.tecla abajotecla arriba
Tecla H: Muestra pista (3er lado).tecla h
Tecla N: Lea el texto en voz.tecla n
![]()
Boton play
![]()
Boton play
![]()
7 Cartas en este set
- Frente
- Atrás
|
FISIOLOGÍA DE LA CORTEZA SUPRARRENAL
|
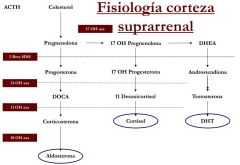
|
|
HIPERPLASIA SUPRARRENAL CONGÉNITA
|
Es una enfermedad hereditaria autosómica recesiva que cursa con una disminución del cortisol y aumento de otras hormonas dependiendo de cual sea el déficit enzimático que haya.
Es de gravedad muy variable y es el tipo más frecuente de enfermedad de hipofunción suprarrenal en el recién nacido |
|
HIPERPLASIA SUPRARRENAL CONGÉNITA POR DÉFICIT DE LA 21 HIDROXILASA
|
No hay paso de progesterona a DOCA.
- No se crea aldosterona - No existe paso de 17OH-progesterona a 17-OH desoxicortisol: no hay cortisol --> no hay feedback negativo--> aumenta la ACHT - Si que existe transformación de androstendiona a testosterona y DHT, y debido al aumento de ACTH existe una DHT muy elevada. Es una forma pierde sal, ya que no hay aldosterona, y es virilizante por el aumento de DHT. CLÍNICA: - Neonatos: virilización en niñas y mayor tamaño en niños. Insuficencia suprarrenal que conlleva: vómitos y deshidratación con hiponatremia e hiperpotasemia que se traduce en una situación de shock y muerte - Infantes: pubertad precoz, pubarquia precoz, aumento de genitales, aumento del desarrollo muscular, crecimiento precoz - Adultos: infertilidad en las mujeres, menstruaciones irregulares, acné, patrón masculino de calvicie... |
|
DIAGNÓSTICO Y TRATAMIENTO DEL DÉFICIT DE 21 HIDROXILASA
|
DIAGNÓSTICO: se realiza mediante un cribado neonatal en el que se mide la cantidad de 17-OH progesterona + sospecha clínica + pruebas (analítica, ecografía y cariotipo y estudio genético)
TRATAMIENTO: a largo plazo debemos sustituir el cortisol y normalizar los andrógenos y a corto plazo evitar la deshidratación y operar la virilización de las niñas (antes de los 2 años) HIDROCORTISONA + FLUDROCORTISONA + NACL (en situaciones de estres triplicar la dosis de hidrocortisona) Para prevenir esto, debemos dar a las embarazadas en las que se sospeche que sus bebés tienen riesgo de tener HSC dexametasona antes de la semana 10. En la semana 12-14 debemos hacer un estudio coriónico y genético y comprobarlo. |
|
DÉFICIT DE LA 11-B HIDROXILASA
|
-Regula el paso de DOCA a aldosterona y de 17-OH desoxicortisol a cortisol.
La clínica es igual a la de la HSC clásica (21OH) pero al estudiarla debemos atender a los niveles de 17-OH desoxicortisol que serán los que estén aumentados. Es menos frecuente que la anterior. El tratamiento consiste en dar hidrocortisona y betabloqueantes para la hipertensión |
|
DÉFICIT DE 3 BETA SDH
|
Impide la transformación de pregnenolona en progesterona, de 17-OH pregnenolona en 17-OH progesterona y de DHEA a androstendiona de forma que no hay ni cortisol, ni aldoserona ni DHT aunque la ACTH está muy elevada.
CLÍNICA: Pérdida de sal y atrofia gonadal. -En niños: feminización (hipogonadismo hipergonadotropo) - En niñas: leve virilización porque DHEA es un andrógeno leve |
|
DÉFICIT 17 OH-HIDROXILASA
|
No permite la transformación de pregnenolona a 17-OH pregnenolona. No hay cortisol ni tampoco DHT, sin embargo la adosterona está muy aumentada: hipertensión y disminución de la renina.
CLÍNICA: -niños: feminización + HTA+ hipogonadismo hipergonadotropo -niñas: genitales normales pero HTA |