- Barajar
ActivarDesactivar
- Alphabetizar
ActivarDesactivar
- Frente Primero
ActivarDesactivar
- Ambos lados
ActivarDesactivar
- Leer
ActivarDesactivar
Leyendo...
Cómo estudiar sus tarjetas
Teclas de Derecha/Izquierda: Navegar entre tarjetas.tecla derechatecla izquierda
Teclas Arriba/Abajo: Colvea la carta entre frente y dorso.tecla abajotecla arriba
Tecla H: Muestra pista (3er lado).tecla h
Tecla N: Lea el texto en voz.tecla n
![]()
Boton play
![]()
Boton play
![]()
102 Cartas en este set
- Frente
- Atrás
- 3er lado (pista)
|
Cuál fue el aporte de Gunter Blobel?
|
Descubrio que las proteínas tienen señales intrínsecas que determinan su transporte y localización en la célula
|
|
|
Cuál fue el aporte de R. Schekman y su equipo?
|
Trabajaron con levaduras y describieron el transporte mediado por Sec12 y Sec17
|
|
|
Función de Sec12
|
Codifica una proteína de membrana que permite que se formen vesículas de secreción
|
|
|
Qué causan las mutaciones en IF2B?
|
Leucoencefalopatía (Ataxia infantil con hipomielinización del SNC)
|
|
|
A qué se debe el síndrome de displasia craniofacial?
|
Mutación en el gen para COPII
|
|
|
Qué es Sar1b?
|
Secretion associated Ras related GTPase 1B, implicada en el transporte de quilomicrones desde el RER hasta Golgi
|
|
|
Qué es la enfermedad de Anderson?
|
Retención de quilomicrones en el intestino por una mutación en la proteína que ayuda a su transporte (Sar1B)
|
|
|
Qué es el SRP?
|
(Signal recognition patern) Que sirve para reconocer el péptido naciente de un ribosoma en el citosol y unirlo a un receptor SRP que posteriormente lo va a unir a un translocon
|
|
|
Qué es un translocon?
|
Canal en la membrana del ER que ayuda a que el peptido ingrese a la luz del reticulo
|
|
|
De qué está constituido el SRP?
|
De seis polipeptidos y una molecula de RNA (7SL)
|
|
|
Qué fosfolípido abunda en la membrana del ER?
|
Fosfatidilcolina (que aumenta en hígados obesos)
|
|
|
Qué es ERGIC?
|
Complejo intermediario entre el RE y el Golgi (también llamado VTC "Vesicular-tubular cluster")
|
|
|
Componentes de COPII
|
Sec12 (actividad GEP), 13 y 23 (actividad GAP), y Sar1-GTP
|
|
|
Qué es Sar1?
|
Proteína guanosina trifosfatasa y monomérica, que regula el ensamble y desensamble vesicular (Es una GTPasa)
|
|
|
Qué es COPII?
|
Complejo de proteínas de cubierta vesicular
|
|
|
Qué son las proteínas SNARE?
|
Proteínas encargadas de facilitar la fusión de vesículas
|
|
|
Función de UDP-N-AcGln
|
Transfiere NAG al aceptor de manosa del dolicol
|
|
|
Qué es el dolicol fosfato?
|
Transportador de la membrana del ER que transporta cadenas de carbohidratos
|
|
|
A qué residuos de aminoácidos en la proteína naciente dentro del ER se transfieren las cadenas de carbohidratos?
|
Residuos de asparagina
|
|
|
Qué producen las mutaciones de subunidades oligosacariltransferasa?
|
Retraso mental
|
|
|
Composicion de la cadena de carbohidratos transferidos al residuos de asparagina?
|
2 NAG
9 Manosas 3 Glucosas |
|
|
Cuál es la función de la oligosacariltransferasa?
|
Transferir el grupo de oligosacaridos desde el dolicol fosfato hasta los residuos de asparagina
|
|
|
Qué son las CDGs?
|
Enfermedades congénitas de glucosidación, síndromes con hipotonia, hipoplasia cerebral y dismorfismo
|
|
|
Qué es CDG1b?
|
Deficiencia de la enzima fosfomanosa isomerasa
|
|
|
Cuál es la función de la fosfomanosa isomerasa?
|
Convertir fructosa 6p en manosa 6p
|
|
|
Cuál es el tratamiento de pacientes con CDG1b?
|
Administración oral de manosa
|
|
|
Función de la glucosidasa II
|
Eliminar la glucosa restante de la glucoproteina que está en el proceso de control de calidad, por lo que esta se va a liberar de la chaperona
|
|
|
Función de la UGGT
|
Reconocer proteínas mal plegadas
|
|
|
Qué son la calnexina y la calreticulina?
|
Chaperonas
|
|
|
Cuál es la importancia de la secuencia ASP-X-SER-THR?
|
Qué cada que aparece se añade una cadena de oligosacaridos
|
|
|
Qué significa ERAD?
|
Degradación asociada a ER
|
|
|
Qué es BiP?
|
Chaperona encargada de "monitorizar" la cantidad de proteínas mal plegadas
|
|
|
Por qué eIFα se fosforila con la activación de sensores de proteínas mal plegadas?
|
Para evitar que se sigan sintetizando proteínas y se eliminen las que están en exceso
|
|
|
Qué es XBP1?
|
Factor de transcripción asociado a estrés en el ER
|
|
|
Función de la PDI
|
Formar enlaces disulfuro mediante la oxidación de SH de residuos de aminoácidos
|
|
|
Qué es UPR?
|
Unfolded protein responses, an "action plan" in case of increased proteins and it includes BiP molecules and Ire1 receptor
|
|
|
En qué parte de la célula sucede la ubiquitinación?
|
Citoplasma
|
|
|
Qué es CFTR?
|
Canal de cloro que sufre mutación en la fibrosis quística
|
|
|
Qué es Ire1?
|
Receptor que se activa por UPR
|
|
|
Qué tipo de proteasoma tenemos los eucariotas?
|
26s
|
|
|
Qué es ATF6?
|
Factor de transcripción activado por proteínas mal plegadas
|
|
|
Qué sucede si muta ERGIC-53?
|
Hay deficiencia de factores de coagulación V y VIII
|
|
|
Qué es la nitisinona?
|
Inhibidor de HPPD que produce ácido homogenistico
|
|
|
En qué porción Golgi se localiza la Alfamanosidasa I?
|
Cis
|
|
|
En qué porción Golgi se localiza la Sialiltransferasa?
|
Trans
|
|
|
Qué es KDEL?
|
Peptido señal que impide que una proteína sea secretada por ER o que bien, una de Golgi se regrese a ER a través de COPI
|
|
|
A qué GTPasa se asocia COPI?
|
ARF
|
|
|
Función de la fosfodiester glucosidasa
|
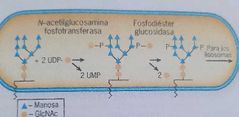
Eliminar los GlcNAc para liberar los p de la cadena de oligosacaridos
|
|
|
Qué son las GGAs?
|
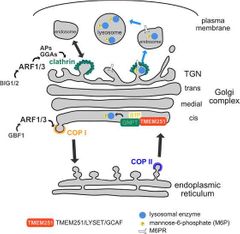
Proteínas adaptadoras presentes en TGN y que se unen a clatrinas y a señales de M-6P en enzimas lisosomales
|
|
|
Función de UDP-N-ACGLN transferasa
|
Transferir grupos P-GLCNAc al oligopeptido
|
|
|
Qué es la mucolipidosis II?
|
Deficiencia de UDP-N-ACGLN TRANSFERASA . Sucede sobre células I. El defecto está en la adición de manosa 6 fosfato.
|
|
|
Qué es la galactosialidosis?
|
Mutación en el gen que codifica para la catepsina A
|
|
|
Qué es el complejo TIM/TOM?
|
Complejo que transloca proteínas a la mitocondria. TIM esta en la membrana interna y TOM en la membrana externa.
|
|
|
Qué sucede en el síndrome de Fanconi?
|
Se excretan sustancias en exceso por los riñones por un error en la proteína α-glucosidasa
|
|
|
Qué sucede en la enfermedad de Gaucher?
|
Defecto en la β-glucosidasa por lo que las proteínas no pueden ingresar a Golgi
|
|
|
Porque Yoshinori recibió un Nobel?
|
Por describir la autofagocitosis
|
|
|
Qué es Rab-GTPasa?
|
Proteínas que forman parte del sistema de transporte y fusion de membranas
|
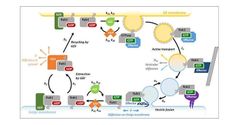
|
|
Qué es el síndrome de Chediak-Higashi?
|
Mutación en la proteína lisosimal LYST que causa hipopigmentacion, deficiencia inmunologica y deficiencia de coagulación
|
|
|
Cuál es la importancia de la proteína NPC1?
|
Que el ébola utiliza esta proteína para empezar a replicarse y también está afectada en individuos con Niemman Pick tipo C por lo que son resistentes a esta enfermedad
|
|
|
Qué es la epistasis?
|
Interacción entre genes mitocondriales y nucleares
|
|
|
Qué es Hsp70?
|
Chaperona que ayuda a que la proteína mitocondrial ingrese a la mitocondria desde el citosol mantenimiendo en parte su estructura
|
|
|
En qué difieren TIM 22 y 23?
|
22 transporta proteínas que se anclan a la membrana lipídica y 23 transporta proteínas que va a la matriz
|
|
|
Qué sucede si muta la proteína de transporte mitocondrial Timp8?
|
Sordera y distonía
|
|
|
Qué es PGC1α?
|
Regulador proteico transcripcional de la biogenesis mitocondrial
|
|
|
Qué tipo de enfermedades puede relacionarse a mitocondrias?
|
Ataxia
Chorea Dystonia Parkinsonismo MELAS MERRF MND |
|
|
Qué son las PTS?
|
Señales de localización peroximal para proteínas
|
|
|
Qué sucede en el síndrome de Zellweger?
|
Hay un problema en la biogenesis peroximal
|
|
|
Qué es la dinamina?
|
Proteína GTPasa responsable de la endocitosis
|
|
|
Qué sucede en la adenoleucodistrofia X?
|
Mutación en transportador ABC (D1) por lo que se acumulan ac. grasos de cadena larga
|
|
|
Qué es la hiperoxaluria?
|
Nefrolitiasis x defecto de alanina-glioxalato aminotransferasa
|
|
|
Qué es la farnesilación proteica?
|
Unión de proteínas a la lámina A con el corte de un grupo farnesil
|
|
|
Límite de tamaño aproximado para la difusión de proteínas a través del poro nuclear
|
40 kDa
|
|
|
Que es NLS?
|
Secuencia rica en AA cerca del extremo carboxilo
|
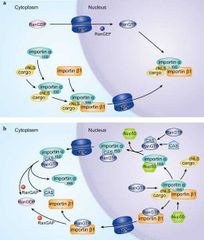
|
|
Que es NES ?
|
Secuencia de aminoácidos rica en leucinas que promueve el transporte fuera del núcleo
|
|
|
De qué depende el transporte de proteínas desde el citosol hasta el núcleo?
|
RanGDP e importinas
|
|
|
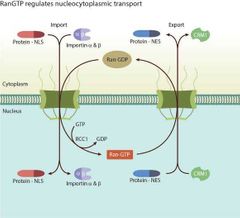
De qué depende el transporte de proteínas desde el núcleo hasta el citosol?
|
Ran GTP y exportinas
|
|
|
De qué depende el transporte de ARNm hacia el citosol?
|
De NXF1 Y NXT1
|
|
|
Cuántas moleculas se importan al núcleo x minuto?
|
60000
|
|
|
En qué consiste la CDG1a?
|
Mutación de la enzima fosfomanosa mutasa que convierte Man-6-P a Man-1-P
|
|
|
En qué consiste la CDGIIB?
|
Deficiencia de glucosidasa 1
|
|
|
Método para diagnosticar CDG
|
Transferrina sérica
|
|
|
Cómo se sintetiza el dolicol?
|
Por farnesil pirofosfato
|
|
|
Cuál es la función de Ire1?
|
Tiene la actividad de splicing de ARNm de XBP1
|
|
|
Función de PERK
|
Kinasa de eIF2α
|
|
|
Función de ATF6
|
Es un factor de transcripción de respuesta a estrés de ER
|
|
|
Chaperonas que asisten plegamiento en matriz mitocondrial
|
Hsp60 y 10
|
|
|
Qué complejo de la cadena respiratoria no se sintetiza en la mitocondria?
|
Complejo 2 (Succinato deshidrogenasa)
|
|
|
Qué son ND1- ND6?
|
Son proteínas del complejo 1 de la mitocondria que se encuentran sus genes en el genoma mitocondrial. A estas proteínas se les unen 39 más que son sintetizadas en el núcleo.
|
|
|
Qué son SDHA-SDHD?
|
Las proteínas que forman el complejo 2 de la mitocondria y que son todas sintetizadas por genoma nuclear
|
|
|
Es la parte (o la proteína )del complejo 3 mitocondrial que es codificado por genoma mitocondrial
|
Apocitocromo b
|
|
|
Qué son las COXI - COXIII?
|
proteínas del complejo 4 mitocondrial que son codificadas por el genoma mitocondrial
|
|
|
Qué son Mfn1 y 2?
|
"Mitofusinas", proteínas implicadas en la fusión de membranas externas mitocondriales
|
|
|
Qué proteínas median la fisión mitocondrial?
|
DLP1 y FIS1
|
|
|
Repaso de los complejos de la cadena respiratoria:
|
Complejo 1 : NADH deshidrogenasa
Complejo 2 : Succinato deshidrogenasa Complejo 3 : Ubiquinol-citocromo c oxidoreductasa Complejo 4 : Citocromo oxidasa Complejo 5 : ATP sintasa |
|
|
Qué Citocromo media la apopotosis mediante la activación de procaspasa 9?
|
Citocromo C
|
|
|
Cuántos ARNt se codifican por el genoma mitocondrial?
|
22
|
|
|
Cuántos ARNr se codifican por el genoma mitocondrial?
|
2
|
|
|
Qué sucede en la enfermedad de Charcot-Marie-Tooth tipo 28?
|
Mutación en Rab7
|
|
|
Qué sucede en la enfermedad de Refsum?
|
Mutación en la enzima fitanoil-Coa hidroxilasa
|
|
|
Qué sucede en la hiperoxaluria primaria?
|
Defecto en la alanina-glioxalato aminotransferasa, se muta una prolina por leucina en el amino terminal
|
|
|
Qué tienen en común las enzimas HRI, PKR, PERK y GNC2?
|
Que fosforilan a eIF2α
|
|
|
Qué sucede en el Síndrome de Vanoshing Withe Matter (VWM)?
|
Muta una isoforma del factor eIF2B
|