- Barajar
ActivarDesactivar
- Alphabetizar
ActivarDesactivar
- Frente Primero
ActivarDesactivar
- Ambos lados
ActivarDesactivar
- Leer
ActivarDesactivar
Leyendo...
Cómo estudiar sus tarjetas
Teclas de Derecha/Izquierda: Navegar entre tarjetas.tecla derechatecla izquierda
Teclas Arriba/Abajo: Colvea la carta entre frente y dorso.tecla abajotecla arriba
Tecla H: Muestra pista (3er lado).tecla h
Tecla N: Lea el texto en voz.tecla n
![]()
Boton play
![]()
Boton play
![]()
170 Cartas en este set
- Frente
- Atrás
|
que es la hematopoyesis
|
SERIE DE EVENTOS CONCATENADOS QUE INICIAN
CON LA AUTOREPLICACION DE LA CÉLULA TALLO TOTIPOTENCIAL, SEGUIDA POR LA DIFERENCIACION Y MADURACION GENERANDOSE ELEMENTOS FORMES SANGUINEOS FUNCIONALES |
|
donde se lleva a cabo la hematopoyesis
|
en la medula osea, especificamente en:
Pelvis 34% • Vértebras 28% • Cráneo y mandíbula 13% • Esternón y costillas 10% • Húmeros, escapulas y clavículas 8% • Fémures 4% |
|
cuales son las células sanguíneas y cuantas se producen al día
|
3x109 eritrocitos por kilogramo de peso
2.5x109 plaquetas por kilogramo de peso 1.0x109 neutrófilos por kilogramo de peso leucocitos |
|
durante la gestación, en que lugares se lleva a cabo la hematopoyesis
|
1. entre la 4ª y 6ª se identifican colonias de blastos en la región aorta/gonada/mesonefros y poco después en el saco vitelino.
2. higado y bazo = 5ta semana 3. medula osea= a partir del 4to mes y durante toda la vida |
|
cual es el precursor común de los linajes endotelial y
hematopoyético |
Hemangioblasto
|
|
que es la diferenciacion
|
secuencia de hechos genéticos que
permiten a una célula sintetizar productos específicos, los cuales le confieren potencialidad para determinada función. POTENCIALIDAD |
|
que es maduracion
|
secuencia de fenómenos bioquímicos y
morfológicos iniciados por la diferenciación y que confieren capacidad funcional a la célula. |
|
a que se refiere un COMPLEJO HETEROGENEO DE CELULAS Y DE SUS RESPECTIVOS PRODUCTOS, NECESARIOS PARA MANTENER Y REGULAR EL CRECIMIENTO DE LA CELULA TALLO
HEMATOPOYETICA |
microambiente hematopoyetico
|
|
cual es la función del microambiente inductivo hematopoyetico
|
libera sustancias capaces de inducir expresión de genes de diferenciación en la CTH
|
|
con que célula inicia la hematopoyesis
|
célula troncal hematopoyética CTH
sigue 2 linajes, mieloide y linfoide |
|
cuale son los marcadores de la celula troncal hematopoyetica
|
Sus marcadores de superficie son CD34,
CD133, CD90 y carecen de marcadores específicos de linaje. |
|
como se dividen los leucocitos
|
-GRANULOCITOS : neutrofilos, eosinofilosy basofilos
-AGRANULOCITOS: monocitos y linfocitos |
|
La CTH se diferencia en:
|
1. de manera estocástica o al azar
2. el MIH es responsable de la selección del linaje celular. |
|
donde descansan las CTH
|
Citoplasma de células reticulares adventicias
|
|
funcion de las celulas estromales
|
son nodrizas de CTH, inhiben la diferenciación mieloide y estimulan la secreción de osteopontina, que activa células T.
|
|
que es el nicho hematopoyetico
|
Microambiente en el que reside la CTH pero con un
enfoque más anatómico que fisiológico. precursor- reconocible morfológicamente progenitor-no reconocible |
|
cual es el sitio ideal para que las CTH realicen sus funciones
|
en el Endostio yace entre el hueso y el sistema
hematopoyético |
|
cuales son los compartimientos hematopoyéticos
|
1.pluripotencial
2.bipotencial 3. unipotencial 4. terminal |
|
cuales son las celulas precursoras de las celulas sanguineas
|
1. Proeritroblasto= eritrocitos
2. monoblasto= monocitos 3. mieloblasto= leucocitos granulocitos 4.megacarioblasto= plaquetas 5. linfoblasto= linfocitos |
|
funcion de la eritropoyetina
|
les ordena a las células madre de la médula ósea producir más glóbulos rojos,
esta en el cromosoma 7 , se produce en riñon e higado |
|
que factor regula la granulopoyesis
|
FACTOR ESTIMULANTE DE COLONIAS GRANULOCITICAS
Estimula actividad quimiotáctica en neutrófilos y monocitos |
|
cual es la funcion de la trombopoyetina
|
estimular crecimiento de los megacariocitos
humanos para producir plaquetas |
|
coagulantes que se emplean en las muestras sanguíneas
|
1.EDTA- cuantificacion de globulos rojos . hb . valorar morfologia
2.citrato sodico- plaquetas y coagulacion 3. heparina - prevenir hemolisis , examenes leucocitarios |
|
cuales son las medidas cuantitativas de glubulos rojos
|
-hemoglobina
-numero de globulos rojos -hematocito |
|
medidas de calidad de los gobulos rojos
|
-VCM
-hemoglobina corpuscular media -concentración media de hemoglobina corpuscular -ancho de distribucion de eritrocitos |
|
se refiere a la proporción del volumen de la sangre
ocupada por los eritrocitos |
hematocrito (htc)
|
|
valores normales de la serie roja
|
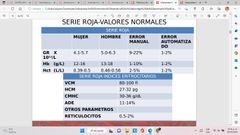
|
|
que es el volumen corpuscular medio (VCM)
|
tamaño promedio de los glóbulos rojos o hematíes de la sangre
|
|
Es el promedio de Hb por eritrocito
|
HEMOGLOBINA CORPUSCULAR MEDIA
(HCM) |
|
que es la CONCENTRACIÓN MEDIA DE
HEMOGLOBINA CORPUSCULAR (CMHC) |
Es el promedio de Hb en un volumen de GR
|
|
que es el ANCHO DE DISTRIBUCIÓN DE ERITROCITOS
(ADE) |
Es la amplitud de la curva de distribución de
los volúmenes de los eritrocitos Es una forma de medir electrónicamente las variaciones de tamaño de GR |
|
que variantes pueden afectarlos resultados en la serie roja
|
-hiperlipidemia
-leucocitosis -cantidad de anticoagulante inadecuado - |
|
cual es la cuenta leucocitaria normal
|
de 4x109 a 11x109 cel/L
|
|
que es la leucocitosis y cual su rango
|
Alto nivel de glóbulos blancos en la sangre
Mayor a 11x109 cel/L Recuerda que al nacer tenemos de 12 a 26 |
|
que es la reaccion leucemoide
|
elevacion extrema de la cuenta de leucocitos a mas de 30, 000 celulas
|
|
que es leucopenia y su valor de referencia
|
Nivel bajo de glóbulos blancos en la sangre
menor a 4x109 cel/L |
|
que es el analisis de la diferencial
|
Es el análisis de los porcentajes relativos de los
diferentes tipos de células de la línea blanca. |
|
a que refiere la neutrofilia
|
Conteo mayor a 7 x 10 9 cel/L de neutrofilos
|
|
causas de neutrofilia aguda
|
1. Uso de glucocorticoides,
catecolaminas, sales de litio – Embarazo y parto –Cirugía – Hemorragias –Crisis convulsivas – Quemaduras – Tabaquismo – Enfermedades inflamatorias – Traumatismos -infecciones bacterias |
|
causas de neutrofilia cronica
|
Neoplasias (Hígado y pulmón)
• Infecciones crónicas (TB miliar) • Fármacos (glucocorticoides, catecolaminas, sales de litio). • Enfermedades inflamatorias crónicas |
|
que es neutropenia y sus causas
|
conteo menor a 1.5 x 10 9 cel/L de neutrofilos
por enfermedades infecciosas e inducida por farmacos (metamizol) , por enfermedad inmunologías como lupus Hepatopatías e hiperesplenismo, leucemia, linfomas |
|
cuales son las causas de eosinofilia
|
-infecciones por helmintos
-Enfermedades alérgicas -neoplasias hemtaologicas |
|
que es basofilia y causas
|
Cuenta mayor de 0.080 x 109/L
– Alergias o inflamación – Reacciones de hipersensibilidad – Colitis ulcerativa – Artritis reumatoide -leucemia mieloide cronica |
|
que es linfocitosis y causas
|
Recuento de linfocitos mayor de 4 x109/L
Neoplasias linfoproliferativas (LLP, LLC) – Mononucleosis infecciosa – Bordetella pertussis – Traumatismos – Cirugías |
|
causas de linfocitopenia y valor
|
• Recuento menor a 1 x 109/L
• Causas – Sepsis bacteriana o fúngica – Glucocorticoides – Neoplasias – Infecciones virales (HIV, tuberculosis) – Linfoma Hodgkin |
|
que es monocitosis y causas
|
Recuento mayor a 0.8 x 109/L
- síndromes mielodisplásicos como Leucemia mieloide aguda - Granulocitopenia crónica de la infancia -– Linfoma Hodgkin -Citomegalovirus y virus varicela zoster |
|
cantidad normal de plaquetas
|
de 150 a 400x109/L
|
|
crietrios para una Púrpura trombocitopénica idiopática
|
aumento de tamaño de plaquetas y disminucion en numero (trombocitopenia)
|
|
cual es el único criterio para diagnosticar anemia y sus valores
|
Hemoglobina
Hb < 13 g/dl en varón adulto • Hb < 12 g/dl en mujer adulta • Hb < 11 g/dl en la mujer embarazada |
|
causas de anemia microcítica hipocrómica (VCM <80 fl)
|
anemia por deficiencia de hierro
por enfermedad cronica talasemias anemia sideroblastica |
|
al mandar a realizar una prueba de hierro serico , cuales son los parametros para diagnosticar una anemia por deficiencia de hierro
|
hierro serico:B
Transferrina: A (transportador) Sat: B Ferritina: B (reserva de hierro) ADE: A PEL: A la biometría la hemoglobina es baja , el vcm y hcm bajo |
|
causas de anemia normocitica (80 a 100 fl)
|
Hiperregenerativa (Reticulocitos altos)
1. Pérdida aguda de sangre 2. Hemólisis 3. hiperesplenismo b) Hiporregenerativa (Reticulocitos bajos) 1. Anemia de enfermedades crónicas 2. Enfermedad renal, hepática, endocrina 3. Médula ósea reemplazada 4.deficiencia de hierro |
|
que son los reticulocitos
|
son glóbulos rojos inmaduros , lo normal es de 0.5% a 2%
|
|
cuales son las causas de anemia macrocítica (VCM >100 fl)
|
anemia megaloblástica (reticulocitos disminuidos)
-deficiencia de vitamina B12 -deficiencia de acido fólico anemia no megaloblásticas -Anemia posthemorragia -alcoholismo -enfermedades metabolicas como hepaticas -anemia hemolitica |
|
como se diagnostica la anemia por deficiencia de vitamina B12
|
-hemoglobina baja
-VCM >100 -retis disminuidos - vitamina B12 < 200 pg/ml |
|
como se diagnostica la anemia por deficiencia de acido folico
|
-hemoglobina baja
-VCM >100 -retis disminuidos -Ac fólico < 2 ng/ml |
|
donde se absorbe el hierro, acido folico y vitamina B12
|
-hierro: duodeno y yeyuno proximal (25 mg/dia)
-acido folico: yeyuno -vit b12:ileon |
|
como puede ser la deficiencia de hierro
|
-ABSOLUTA: las reservas de hierro son bajas o agotadas
-RELATIVA: reservas de hierro normales o aumentadas, Suministro de hierro a la medula ósea es inadecuado |
|
cual es la función de la hepdicina
|
es una hormona producida por el hígado,
Regulador metabolico del FE, evitar así la sobrecarga de Fe y su toxicidad El ejecutor de la Hp es la Ferroportina (FP), único exportador de Fe |
|
mutación que causa Anemia por
resistencia al hierro |
Mutación TMPRSS6 que causa Producción
alta de hepcidina y se bloquea la absorción de hierro |
|
signos y síntomas de anemia por deficiencia de hierro
|
-fatiga y debilidad
-dolor de cabeza, pica, sindrome de piernas inquietas, disnea de esfuerzo -palidez, piel seca, coiloniquia , cabello seco |
|
cual es el tratamiento de la anemia por deficiencia de hierro
|
inicio con hierro oral Dosis diaria recomendada: 100-200mg
si no funciona por intolerancia , necesidad de recuperación rapida o fracaso oral poner hierro IV |
|
cual es el objetivo del tratamiento oral de hierro
|
Mejorar las reservas de hierro y Ferritina ≥100µg/l en 3 meses
|
|
de que manera se absorbe el acido fólico
|
como monoglutamato
poliglutamato en alimentos |
|
cual es la forma mas abundante y activa del acido folico
|
tetrahidrofolato
|
|
cuanto requerimiento se necesita de acido folico al dia y cuanto es su reserva
|
en adultos es de 400 microgramos pero con 50 esta bien y su reserva es de 5 mg
|
|
cuales son las funciones del acido folico
|
-sintesis de timidilato : forma timina para la producción de DNA
-síntesis de metionina |
|
es una nota
|
en la anemia megalobalstia hay nucleos inmaduros en el microscopio
|
|
en que forma esta constituida la molecula de vitamina B12 (cobalamina) y cuales son sus formas activas en el ser humano
|
-grupo corrina
-benzimidazol -radical variable : por eso tiene diferentes nombres formas activas: adenosilcobalamina y metilcobalamina cianocobalamina: versión que compramos |
|
cual es el requerimiento diario de vitamina b12
|
es de 2 microgramos , su reserva es de 2-5 mg y dura de 3 a 4 años
|
|
de que forma se absorbe la cobalamina
|
complejo colabamina/fI
la sustancia R o transcobalamina 1 hace que no se desnaturalice en el estomago |
|
cierto o falso: si hay deficiencia de vitamina b12 no hay acido fólico funcional
|
cierto
|
|
a que se debe la anemia megaloblastica
|
es debido a que no hay suficiente acido fólico que produce DNA por consecuencia hay una eritropoyesis ineficaz
se acompaña de anemia , polisegementacion de neutrofilos y trastornos de maduración eritroblastica, trombocitopenia y leucocitopenia |
|
cuales son las causas de deficencia de vitamina B12 y acido folico
|
de b12 por alteraciones en la absorcion y de acido folico por falta de ingesta (se encuentra principalmente en carnes)
|
|
como puedes diferenciar una anemia por deficiencia de b12 o acido folico en sus manifestaciones clínicas
|
en ambas hay anemia , palidez/ictericia, glositis atrófica de Hunter, hiperpigmentación ungueal.
en falta de cobalamina hay sintomas neurologicos como pérdida de la sensación de posición en el segundo dedo del pie, es util para la produccion de mielina y osteoporosis |
|
como se diagnostican las anemias megaloblasticas en la biometria hematica
|
Macrocitosis leve (100-105 fL), moderada (106-115 fL) e intensa (>116 fL)
Aumento de ADE (ancho de distribución eritrocitario) Bicitopenia o pancitopenia Granulos gruesos Reticulocitopenia polisegementacion de los neutrófilos |
|
en la quimica sanguinea que valores estarian aumentados en las anemias megaloblasticas
|
bilirrubina indirecta y deshidrogenasa láctica (DHL)
|
|
que valores de cobalamina y folato serico son los diagnósticos para anemia megaloblástica
|
VB12- <200 pg/ML
AF-<2 ng/mL |
|
cual es el tratamiento para la anemia megaloblastica
|
ÁCIDO FÓLICO 1 a 5 mg/día VO 3 a 4 meses
1000 μcg IM diarios/1 semana, 1000 μcg semanarios por 4 semanas |
|
que es lo que provoca las interleucinas y hepcidina en las anemias por padecimiento cronico
|
las interleucinas disminuyen la producción de eritropoyetina , decrementa la produccion de eritrocitos y de sus precursores
-hepcidina + - evita su funcion , por lo que no se absorbe hierro |
|
cual es la principal cusa de anemia
|
deficiencia de hierro
|
|
cuadro clinico de anemias por padecimiento cronico
|
-los de su patologia
-puede haber anemias NN o microcitica hipocromica, lo que la diferencia de la DH es que aqui la ferritina es normal o incrementada |
|
que es la anemia aplasica o insuficiencia de la medula osea
|
presencia de citopenias (mono,bi o pancitopenia, es decir, 1, 2 o las 3 series sanguineas disminuyen ) debido a la sustitución de las células hematopoyeticas de medula ósea por grasa
|
|
cual es la fisiopatología de la anemia aplásica
|
por formación de anticuerpos que destruyen las celulas , por ALTERACION EN LA CELULA TALLO
|
|
cual es la etiologia de la anemia aplasica
|
-PRIMARIA O IDIOPATICA- por drogas, quimicos, virus,
-SECUNDARIA - metamizol y metimazol , Hemoglobinuria paroxística nocturna |
|
sindromes presentes en la anemia aplasica
|
-sindrome anemico
-sindrome febril -sindrome hemorragico |
|
que encontraras en una biometría hematica con anemia aplasica y criterios para que sea severa y muy severa
|
-pancitopenia (todas las celulas bajas)
-se debe realizar una biopsia de medula osea neutrofilos <500 plaquetas <20,000 reticulocitos corregidos <1% celularidad <25% (2 a 3 criterios + celularidad) muy severa < 200 neutrofilos |
|
tratamiento de la anemia aplasica
|
-inmunosupresores
-transfundir plaquetas -trasplante alogenico |
|
que son las anemias hemoliticas
NOTA: anemias megaloblasticas tambien el eritrocito des destriuye antes de los 120 dias |
afección sanguínea que ocurre cuando los glóbulos rojos se destruyen más rápido de lo que se pueden reemplazar e igual no se producen
|
|
clinica las anemias hemoliticas
|
-disminución de los glóbulos rojos
-ictericia - debido al + de bilirrubina indirecta y DHL -esplenomegalia -anemia megalobalstica |
|
cuales son los tipos de hemolisis y sus diferencias
|
-extravascular- se lleva dentro del macrofago la destruccion del eritrocito
-intravascular- el eritrocito se destruye en la circulacion , es mas grave en la instravascular hay coloración rojiza de la orina (hemoglobinuria) + disminución de los glóbulos rojos -ictericia y esplenomegalia , anemia megaloblastica |
|
en un aspirado de medula osea en anemia hemolitica que encontrarías
|
Hiperplasia de serie roja en médula ósea
■ Normoblastos en sangre periférica ■ Reticulocitosis ■ Macrocitosis |
|
cuales son los tipos de anemia hemolitica congenita
|
MEMBRANOPATIAS: esferocitosis hereditaria
ENZIMOPATIAS: via de las pentosas y via EMH HEMOGLOBINOPATIAS: talasemias y drepanocitosis NOTA: hay adquiridas que son autoinmunes y Hemoglobinuria paroxística nocturna |
|
en la Hemoglobinuria paroxística nocturna que receptores disminuyen
|
disminuye la cantidad de cd55 y cd59
|
|
membranopatia mas importante en la anemia hemolitica congenita
|
esferocitosis hereditaria en donde hay defectos en proteínas de la membrana del eritrocito y hace que se destruya antes
|
|
cual es el defecto mas común de la membrana del eritrocito que produce esferosis hereditaria
|
proteina ankirina
|
|
cual es el tratamiento de la esferocitosis hereditaria
NOTA : para su diagnostico es Fragilidad osmótica |
Suplementos de ácido fólico (5 mg/dia)
y esplenectomia |
|
que son las talasemias
|
ANORMALIDADES HEREDITARIAS DE LA HEMOGLOBINA,CARACTERIZADAS POR LA PRODUCCION INSUFICIENTE DE UNA O MAS CADENAS DE GLOBINA
|
|
cuales son los tipos de talasemias
|
- beta talasemias - alterado cromosoma 11 , EXCESO RELATIVO DE CADENAS α de la hb
-alfa talasemias . alterado cromosoma 16 |
|
cual es el diagnostico de la talasemia beta
|
-anemia microcitica hipocromica , sin datos de falta de hierro
-historia familiar -esplenomegalia GOLD STANDAR- electroforesis de hemoglobina |
|
que es la drepanocitosis o anemia por células falciformes
|
Es causado por herencia homocigota de genes para la hemoglobina S. Los eritrocitos en forma de hoz ocluyen el vaso y son propensos a la hemólisis
NOTA: se diagnostica por electroforesis de hemoglobina |
|
que es la anemia hemolítica autoinmune y cual es su tratamiento
|
es en la que el eritrocito es destruido por actividad inmune contra sus antígenos de membrana, es decir, hay anticuerpos que destruyen los eritrocitos
se trata con esteroides , inmunodepresores |
|
cuales son las vias enzimaticas que intervienen en la correcto desarrollo del eritrocito
|
via hexosa monofosfato - falla la glucosa 6 fosfato
via de glicolisis anaerobia - falla el piruvato cinasa |
|
para que sirve la prueba de coombs indirecto
|
detectar la enfermedad hemolítica del recién nacido , se realiza en la madre
|
|
para que sirve la prueba de coombs directa
|
para detectar una enfermedad hemolítica autoinmune , detecta los anticuerpos sobre la superficie de los eritrocitos
|
|
que es la HEMOGLOBINURIA PAROXISTICA
NOCTURNA |
TRASTORNO CLONAL DE LA CELULA TALLO
HEMATOPOIETICA, ADQUIRIDO, CARACTERIZADO POR UNA MAYOR SENSIBILIDAD DE TODOS LOS ELEMENTOS FORMES DE LA SANGRE A LA ACCION DEL COMPLEMENTO Y RELACIONADO CON MUTACIONES EN EL GEN PIG-A se clasifica en clasica, subclinica y falla de medula osea |
|
cuales proteinas estan reducidad o ausentes en la HEMOGLOBINURIA PAROXISTICA
NOCTURNA |
la CD55 Y CD59, importante proteína reguladora del complemento, en la superficie de las células sanguíneas. Como consecuencia, las células son susceptibles a la activación del complemento, lo que conduce a la hemólisis intravascular continua de los eritrocitos.
|
|
manifestaciones clinicas de la HEMOGLOBINURIA PAROXISTICA
NOCTURNA |
leucopenia, la trombocitopenia, las trombosis arteriales y venosas, hemoglobinuria
NOTA: tratamiento con eculizimab , esteroides |
|
que son las neoplasias mielodisplasicas
Displasia: Alteración en la maduración de las células |
Son proliferaciones clonales de células germinales
medulares multipotentes que retienen la capacidad de diferenciarse hasta estadios maduros, pero lo hacen de forma defectuosa e ineficaz, resultando células deficientes en número, función y con morfología alterada, con riesgo variable de transformación leucémica AGUDA En pocas palabras , es cuando las celulas sanguineas no maduran ya que la medula osea las fabrica de forma anomala |
|
cuales son los criterios diagnosticos de SMD
|
Citopenia periférica NO atribuida a enfermedad detectable
• Cambios displásicos en al menos UNA línea celular • Anormalidades citogenéticas ADQUIRIDAS en células hematopoyéticas • Incremento de blastos, mínimo 5% SE NECESITAN MINIMO 2 |
|
que alteraciones en las celulas sanguineas puede haber en los SMD
|
ERITROIDE : megaloblastosis, multinucleados
MEGACARIOCITA: multinucleados GLANUCLOCITOS : Pseudo Pelger (neutrofilos tiene 2 segmentos no 3) y Hipogranulaciòn |
|
cual es la patpgenia de los SDM
|
lo que sucede es que hay apoptosis disminuida , es decir, las celulas viven mas por lo que hay proliferacion aumentada y estas son mas propensas a cambios anormales
lo causa quimioterapia y radiación sus manifestaciones son anemia, infecciones y hemorragias |
|
como diagnosticar los SDM
|
aspirado de medula osea con biopsia de hueso y examen de genes
|
|
como se dividen las neoplasias linfoides
|
Linfomas
Leucemia Aguda Linfoblástica Leucemia Linfocítica Crónica |
|
como se dividen las neoplasias mieloides
|
Leucemia Aguda Mieloblástica
Leucemia Mieloide Crónica |
|
que es la leucemia linfoblastica aguda
|
enfermedad clonal maligna de células
precursoras linfoides en distintos grados de maduración que da lugar a invasión de la médula ósea en >20% de la celularidad total principal en niños y México |
|
caracteristicas de la leucemia linfoblastica aguda
|
• pérdida de control de la
proliferación, diferenciación e inhibición de la apoptosis de la línea linfoide hay 3 tipos: L1:es la más uniforme de todas y la menos indiferenciada, con células de tamaño pequeño y escaso citoplasma L2 - linfoblastos de tamaño variable, nucléolos más evidentes y menos diferenciados L3- tipo Burkitt) se caracteriza por células grandes, indiferenciadas, con nucléolos evidentes y numerosas vacuolas |
|
que cromosoma es el afectado en la leucemia linfoblastica aguda
|
cromosoma philadelphia, produce la proteina BCR-ABL
|
|
manifestaciones clinicas de la leucemia linfoblastica aguda
|
Inicio súbito (generalmente menor de 4 semanas)
▪ Síndrome anémico, síndrome febril, síndrome hemorrágico, síndrome infiltrativo (adenomegalias, hepato y esplenomegalia) |
|
que se encuentra en los estudios de laboratorio de la leucemia linfoblastica aguda
|
CH: Anemia/Leucopenia o leucocitosis/Plaquetopenia, regularmente pancitopenia
•Hiperuricemia •DHL elevada |
|
que es la anemia linfocitica cronica
|
neoplasia compuesta de linfocitos B monomórficos, pequeños, redondos que infiltra la sangre periférica, la médula ósea, el bazo y los ganglios linfáticos.
Junto con el infiltrado previo se mezclan prolinfocitos y parainmunoblastos, formando centros de proliferación en los infiltrados hísticos se caracteriza por la proliferación y acumulación de linfocitos de aspecto maduro en la médula ósea, la sangre, los ganglios linfáticos y el bazo. es la mas comun en adultos |
|
diagnostico para la leucemia linfocítica crónica
|
Edad mayor de 60 años
Linfocitosis por encima de 5000x109/L -trombocitopenia , anemia -Hepatomegalia y esplenomegalia -síntomas como pérdida de peso > 10% en últimos 6 meses • Fatiga • Fiebre > 38° C por 2 o más semanas • Diaforesis nocturna > 1 mes -GOLD ESTANDAR- inmunofenotipo --alteraciones cromosomicas= del ( 13 q14 ) o cromosoma 17 -tp53(regula genoma humano) |
|
tratamiento de la leucemia linfocitica cronica
|
-Ibrutinib
-trasplante autólogo |
|
que es la leucemia mieloblastica aguda
|
Enfermedad maligna que resulta de
la expansión clonal de blastos mieloides en la sangre periférica, médula ósea o en otros tejidos. la palabra mieloblástica denota su origen a partir de los mielocitos, células inmaduras que normalmente se convierten en eritrocitos, leucocitos plaquetas maduras mas comun en adultos >64 años |
|
cuales es la clasificación morfológicas de la leucemia mieloblastica aguda
|
M0= LMA con mínima diferenciación
M1=LMA sin maduración M2= LMA con maduración M4= LMA mielomonoblástica M5a= LMA monoblastica pura M5b= LAM monocitica pura M6= LAM eritroleucemia la M3= Leucemia promielocítica aguda, es urgencia , ya que hay hemorragias m7= LMA megacarioblastica, trisomía 21 |
|
que es la leucemia aguda
|
proliferación neoplásica de cualquier célula del tejido hematopoyético, la célula anormal en la leucemia aguda es el blasto
|
|
clasifiacion de la OMS de la leucemia mieloblastica aguda
|
-LMA con anormalidades genéticas recurrentes
-LMA con cambios asociados a mielodisplasia - Neoplasias mieloides asociadas a tratamiento - LMA, no especificadas en otro sitio -Sarcoma mieloide - Proliferaciones mieloides asociadas a síndrome de Down |
|
manifestaciones clínicas de la leucemia mieloblastica aguda
|
-Síndrome anémico
•Síndrome infiltrativo (encías, organomegalias abdominales ) •Síndrome febril •Síndrome hemorrágico -Palidez •Hipertrofia gingival •Hepato y esplenomegalia •Hipertermia •Púrpura seca y húmeda |
|
diagnostico de la leucemia mieloblastica aguda
|
•CH: Anemia/Leucopenia o leucocitosis/Plaquetopenia
•Hiperuricemia •DHL elevada •TP, TTPa, TT alargados, hipofibrinogenemia -en el frotis hay bastones de Aüer y blastos con granulación citoplásmicade , casi siempre de la M3 |
|
que es la leucemia mielode cronica
|
Ateraciones CLONALES HEMATOPOYÉTICAS que surgen de la transformación de la célula madre CD34+.
• La principal característica es la sobreproducción de células maduras |
|
cuales son las neoplasias mieloproliferativas crónicas
|
-leucemia miolode cronica
-policitemia vera -trombocitopenia esencial -mielofibrosis idiopatica |
|
leucemia cronica mas frecuente en México
|
leucemia mieloide crónica
media de 45-65 años |
|
cromosma involucrado en la LMC
|
El cromosoma (Ph+) es la translocación balanceada del segmento ABL del cromosoma 9 y el segmento BCR del cromosoma 22
|
|
etapas de la LCM
|
-fase cronica- mayor diagnostico
-fase acelerada -fase blastica |
|
estándar de oro para el diagnostico de la LMC
|
es un PCR , análisis citogenético ( incluyendo el FISH: fluorescence in situ hybridization)
|
|
tratamiento para la LCM
|
trasplante de progenitores hematopoyéticos es en donador HLA-compatible (trasplante alogénico)
-inhibidores de la tirosin-quinasa como nilotinib y dasatinib |
|
cual es la a anemia hemolítica hereditaria más frecuente en México
|
esferocitosis hereditaria, es cuando los glóbulos son mas pequeños (esferocitos) y tienen mucha hemoglobina
|
|
como se diagnostica la esferocitosis hereditaria
|
-Pacientes que presenten CHCM >36 gr/dL, sospechar de esferocitosis hereditaria.
- prueba de fragilidad osmótica con cloruro de sodio con sangre incubada -reticulocitosis e hiperbilirrubinemia indirecta |
|
se refiere a una normalidad en tamaño de un
ganglio linfático causada por invasión o propagación de células inflamatorias o neoplásicas. |
linfadenopatia
son principalmente localizadas , se da en cabeza cuello, inguinal y supraclavicular prinicipalmente |
|
causas de linfadenopatia
|
1-procesos infecciosos -virus, bacterias, hongos
2-procesos inflamatorios 3-enfermedades autoinmunes 4-enfermedades hematologicas como los linfomas |
|
cuales son las indicaciones para realizar una biopsia ganglionar
|
-Crecimiento ganglionar después de 3 semanas de
estudio sin diagnóstico etiológico. • Falta de disminución de tamaño después de seguimiento de 4 a 6 semanas. • Falta de regresión a tamaño considerado normal para la edad del paciente y la localización, en 10 a 12 semanas. • Localización cervical baja o supraclavicular |
|
caracteristicas demograficas del linfoma de Hodgkin
|
-Se da principalmente en mayores de 65 años
-masculinos NOTA: en los pacientes pediatricos la leucemia linfoblastica aguda , neuroblastomas y linfomas Hodgkin y no Hodgkin - |
|
que es un linfoma
|
Transformación neoplásica de células linfoides, las cuales se encuentran generalmente en el tejido linfoide secundario (ganglionar y extraganglionar como el bazo, placas de peyer
NOTA: tejido linfoide primaria- medula osea, timo |
|
cuales son los principales agentes biológicos que se relaciones con los linfomas
|
-HTLV-1
-epstein Barr -citomegalovirus -helycobacter pylory |
|
clasificacion de los linfomas Hodgkin
|
-clásico : hay 4 subtipos histologicos que son
1- esclerosis nodular 2-celularidad mixta - mas comun en México , por EBV -rico en linfocitos -deplecion linfoide células de Reed-Sternberg en analisis morfologico , ojos de buho , expresa Cd15 y dc30 -no clásico : 5%. recibe el nombre de predominio linfocitico nodular, su célula definitoria es la L y H o pop corns cell , expresa cd20 |
|
como se divide el linfoma de no Hodgkin
|
-neoplasias de celulas B = 80%, el mas frecuente es el linfoma difuso de celulas grandes de tipo B
-neoplasias de celulas T = el mas comun es el de celulas T perifericas no especificado |
|
diferencias entre la presentacion clinica de los linfomas de Hodgkin y no Hodgkin
|
-ganglionar- los Hodgkin
-extraganglionar - los no Hodgkin Presencia de síntomas “B”: fiebre, diaforesis, pérdida de peso |
|
a que se refiere la clasificacion de Ann arbor
|
para saber que tan extendidos estan los linfomas en el cuerpo , hay 4 estadios
|
|
diagnostico necesario para un linfoma
|
-biopsia del ganglio escisional o incisional
-inmunohistoquímico - el estudio de imagen es el PET-CT |
|
tratamiento del linfoma de Hodgkin
|
-quimioterapia con el esquema ABVD
-anticuerpos monoclonales |
|
tratamiento del linfoma de no Hodgkin
|
C: Ciclofosfamida
H: Hidroxi-Adriamicina O: Vincristina (Oncovin) P: Prednisona R: Rituximab - solo linfoma B |
|
que es el mieloma multiple
|
enfermedad en donde se acumulan celulas plasmaticas , que son celulas B
•Dicha clona secreta una inmunoglobulina (Ig) monoclonal (paraproteína o proteína M). •La presencia de esta proteína M se conoce como gammopatía monoclonal |
|
que organos afecta el mieloma multiple y manifestaciones clinicas
|
hueso y riñon
Manifestaciones Clínicas. C hiperCalcemia R falla Renal A Anemia normocitica normocromica B Lesiones óseas (Bone) -Nivel de proteinas M>30 g/L -% de celulas plasmaticas en la medula osea= >10% |
|
interleucina mas importante en el mieloma multiple
|
es la 6
la celulas no mueren , ya que inbibe la aopotosis |
|
tratamiento para mieloma multiple
|
-inhibidor de proteasoma
-corticoides -inmunomodulador -bifosfonatos y RT para los huesos |
|
clasificación de las neoplasias mioeloproliferativas crónicas
|
LMC BCR-ABL1 positiva
LEUCEMIA NEUTROFÍLICA CRÓNICA POLICITEMIA VERA TROMBOCITEMIA ESENCIAL MIELOFIBROSIS PRIMARIA LEUCEMIA EOSINOFÍLICA CRÓNICA NO ESPECIFICADA EN OTRA PARTE NMC, NO CLASIFICABLE |
|
que expresa el BCR-ABL
|
leucemia mieloide cronica
|
|
donde se expresa el JAK2
|
1-mielofibrosis 57%
2-policitemia vera 99% 3- trombocitemia esencial 72% |
|
que es policitemia vera
|
NMPC caracterizada por incremento en la producción eritrocitaria independiente de los
mecanismos de regulación habitual de la eritropoyesis. Mutación de ganancia de función casi universal: JAK2 V617F - receptor de la eritropoyetina Cursa con panmielosis - las 3 lineas sanguíneas se incrementan |
|
fases de la policitemia vera
|
-fase cronica
-fase acelerada -fase blastica o gastada |
|
manifestaciones clinicas de la policitemia vera
|
-trombosis y hemorragias
-sindrome de hiperviscosidad - se duerme , cefalea, vision borrosa , pulpejo de los dedos duelen , prurito cuando se bañan , ulceras peptica , dispepsia SIGNOS - pletorico-rojo -hiperemia conjuntival -esplenomegalia |
|
diagnostico de la policitemia vera
|
CRITERIOS MAYORES
Hb > 16.5 g/dL hombres, > 16 g/dL mujeres o elevación del Hto (> 49% hombre; > 48% mujer) o incremento de la masa eritrocitaria (>25% arriba del valor predicho normal) Biopsia de hueso: hipercelularidad + panmielosis (crecimiento trilineal) con proliferación prominente eritroide, granulocitaria y megacariocítica con megacariocitos pleomórficos maduros (diferentes en tamaño) -JAK2 V617F o mutación del exón 12 del JAK2 CRITERIOS MENORES Niveles de EPO sérica bajo 2 mayores y un menor o los 3 mayores |
|
gold standar en el tratamiento de policitemia vera
|
Ruxolitinib
|
|
que es la trombocitenia esencial
|
NMPC que implica proliferación de
megacariocitos (grandes y maduros) en la médula ósea y clínicamente por trombosis y/o hemorragia Mutación JAK2 V617F, receptor MPL Cursa con proliferación megacariocítica , no hay ni aumento de leucocitos ni Hb |
|
manifestaciones de la trombocitenia esencial
|
hay hemorrágicas, trombosis eritromelalgia, purpura seca o humeda
|
|
tratamiento de la trombocitenia esencial
|
-aspirina
-interferon |
|
que es la MIELOFIBROSIS PRIMARIA
|
NMPC caracterizada por proliferación de megacariocitos y granulocitos, asociada a depósito
reactivo de tejido conectivo fibroso en MO y hematopoyesis extramedular, es decir la hematopoyesis se va a higado y bazo Mutación es la JAK2 V617F Cursa con proliferación megacariocítica y mieloide, disminución de la eritropoyesis y depósito de fibras reticulínicas |
|
laboratorio de biometria hemtaica de la MIELOFIBROSIS PRIMARIA
|
leucocitosis , trombocitosis y anemia , leucoeritroblastosis,
aumento de DHL |
|
manifestaciones clinicas de la MIELOFIBROSIS PRIMARIA
|
-anemia
-hipermetabolismo como diaforesis, fiebre , baja de peso -esplenomegalia -hepatomegalia |
|
tratamiento de la MIELOFIBROSIS PRIMARIA
|
-PRIMERA LÍNEA: Ruxolitinib
-esplenectomia -trasplante alogénico |