- Barajar
ActivarDesactivar
- Alphabetizar
ActivarDesactivar
- Frente Primero
ActivarDesactivar
- Ambos lados
ActivarDesactivar
- Leer
ActivarDesactivar
Leyendo...
Cómo estudiar sus tarjetas
Teclas de Derecha/Izquierda: Navegar entre tarjetas.tecla derechatecla izquierda
Teclas Arriba/Abajo: Colvea la carta entre frente y dorso.tecla abajotecla arriba
Tecla H: Muestra pista (3er lado).tecla h
Tecla N: Lea el texto en voz.tecla n
![]()
Boton play
![]()
Boton play
![]()
29 Cartas en este set
- Frente
- Atrás
|
CARACTERÍSTICAS DE LOS TUMORES INFANTILES
|
Las células tumorales son embrionarias o sarcomas.
Las localizaciones anatómicas son más profundas, los diagnósticos suelen ser accidentales (palpación de masa) o tardíos (evolucionados). |
|
TUMORES SÓLIDOS MÁS FRECUENTES EN NIÑOS
|
T.del SNC ( 15-25%)
Neuroblastoma (7-10%) T. de Wilms (6%) Retinoblastoma (3%) Sarcomas: -Rabdomiosarcoma (5%) -Osteosarcoma (4%) -Sarcoma de Ewing (3%) |
|
NEUROBLASTOMA
|
Es el tumor sólido extracraneal más frecuente en la infancia y tiene su origen en la cresta neural (ganglios simpáticos pararaquídeos y suprarrenales).
Asienta sobre células embrionarias que pierden su diferenciación y continúan su división produciendo neuroblastos inmaduros. - TUMORES MADUROS Y BENIGNOS: ganglioneuroma y feocromocitoma - TUMORES INMADUROS Y MALIGNOS: neuroblastoma y ganglioneuroblastoma. Estos tumores representan el 7-10% de los tumores infantiles y son más frecuentes en niños menores de 5 años. La localización es abdominal en el 75% de los casos y torácica en el 20% (mediastino posterior) |
|
CLÍNICA DEL NEUROBLASTOMA
|
-Suele ser una masa abdominal indolora que rebasa la línea media.
-Tiene también localización cervico-torácica, da pocos síntomas aunque provoca compresión raquídea o síndrome de vena cava superior y síndrome de claude bernard horner. - Forma extendida: da síndrome constitucional - Nódulos azulados, frecuente en el lactante, y mejor pronóstico que el resto (*) Ojos de mapache por infiltración retrobulbar |
|
CLÍNICA POR APARATOS
|
- Área paraespinal: son pacientes con NB en reloj de arena que se extiende al espacio intramedular y comprimen la médula. Pueden referir dolor de espalda, cojera, hipotonía, arreflexia, atrofia muscular, paraplejia, escoliosis o alteración de esfínteres.
- En la piel: las metástasis subcutáneas (estadio 4S) se manifiestan como nódulos duros, no dolorosos, azulados y frecuentes en lactantes. - Síntomas paraneoplásicos: al ser tumores productores de catecolaminas pueden presentar sofocos, cefaleas, palpitaciones, diarrea con fallo del medro asociada a distensión abdominal, hipertensión por aumento de renina, en los que existe compresión renovascular. También pueden producir parathormona y producir una hipercalcemia. Puede secretar péptido intestinal vasoactivo dando lugar a diarrea acuosa intratable. |
|
SÍNDROME OPSOCEREBELOMIOCLÓNICO
|
Consiste en descargas bruscas de movimientos rápidos involuntarios de los ojos en todas las direcciones de la mirada, que no fija (opsoclonus). Asociadas a ellas presenta también mioclonías de músculos del tronco y las piernas. Estos síntomas son paraneoplásicos y se pueden resolver o no con la resección tumoral. Van asociadas a buen pronóstico vital, pero la mejoría puede ser lenta y parcial, con frecuencia es necesario tratar los síntomas.
|
|
FORMA 4S DEL LACTANTE
|
Es un tumor primario localizado, diseminado al hígado, piel y MO con menos del 10% de células tumorales.
Aparece en niños de menos de un año de edad y MIBG da negativa en MO. |
|
DIAGNÓSTICO DEL NEUROBLASTOMA
|
Enolasa neuronal especifica elevada
Ac. VM, Ac HV, catecolaminas, dopamina elevadas en orina. Su grado de elevación esta asociada a supervivencia acortada Tirosin-hidroxilasa elevada (marcador de enfermedad residual y recidivas muy precoz.) Aumento LDH y ferritina Rx tórax: masa mediastino posterior, masa retrocardiaca- abdominal en reloj de arena Ecografía/ RMN Gammagrafia con MIBG Gammagrafia con Tc Biopsia-reseccion quirúrgica: histología, (N-Myc, citogenética, Índice de ADN) Medula Osea bicrestal |
|
BIOLOGÍA MOLECULAR DEL NEUROBLASTOMA
|
- Amplificación del gen N-Myc (NMA): se asocia a mal pronóstico por rápida progresión tumoral.
- Delección de 1p: se asocia casi siempre con NMA. Si aparece una presentación aislada, conlleva un mayor riesgo de recidiva local. - Alteraciones de 11q: se encuentra en niños mayores y no se asocia con NMA. El pronóstico es desfavorable. - Índice de DNA: hipoploidia es signo de mal pronóstico. |
|
ESTADIOS
|
1. Localizado con excisión macro completa con o sin enfermedad residual microscópica y sin ganglios positivos.
2. Localizado sin excisión macro completa pero sin ganglios positivos. 2B. Localizado con/sin excisión macro completa y ganglios positivos ipsilaterales pero negativos los contralaterales. 3. Tumor irresecable unilateral que pasa la línea media, con ganglios positivos/negativos regionales ó tumor unilateral localizado con ganglios postivos contralaterales ó tumor irresecable con infiltración bilateral y afectación de ganglios. 4. diseminado 4S. piel hígado y médula ósea. |
|
GRUPO PRONÓSTICO Y TRATAMIENTO
|
BAJO RIESGO: cirugía (98%)
RIESGO INTERMEDIO: cirugía y quimioterapia (90-95%) ALTO RIESGO: cirugía + quimioterapia + megaterapia + radioterapia+ ácido 13 cisretinoico + ac monoclonales antigangliosidos (40-50% supervivencia9 |
|
DIAGRAMA DE ESTUDIO DEL NEUROBLASTOMA
|
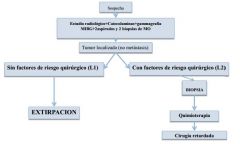
|
|
TUMOR DE WILMS (NEFROBLASTOMA)
|
Son tumores derivados del blastoma renal embrionario. Es el tumor renal más frecuente en la infancia y su frecuencia es del 6% dentro de los cánceres infantiles.
Tiene su pico de incidencia entre los 3 y 4 años y la incidencia es igual en ambos sexos. |
|
BIOLOGÍA MOLECULAR DEL TUMOR DE WILMS
|
Este tumor está causado por la alteración en los genes responsables del desarrollo genito-urinario. Las alteraciones moleculares que producen la inactivación del gen WT1 o la pérdida del marcaje del gen WT2 pueden provocar la persistencia de restos nefrogénicos que se pueden transformar en el tumor de wilms:
- TIPO 1: presencia de restos nefrogénicos intralobares, edad de presentación precoz, histología favorable, predominio de estroma y aniridia. Anomalías genitourinarias: WARG y Denys-Drash - TIPO 2: presencia de restos nefrogénicos perilobares, edad de presentación tardía, histología con anaplasia o favorable con blastema. Síndromes con gigantismo (síndrome de Beckwith-wiedeman) |
|
SÍNDROME DE WARG
|
wilms + aniridia + malformación genitourinaria + retraso mental
|
|
MALFORMACIÓN DE DENYS-DRASH
|
Pseudohermafroditismo + enfermedad renal degenerativa + tumor de wilms
|
|
SÍNDROME DE BECKWITH- WIEDEMAN
|
Exoglosia + gigantismo + onfalocele + hemihipertrofia + visceromegalias + tumor de wilms
|
|
CLÍNICA DEL TUMOR DE WILMS
|
Masa abdominal palpable, indolora, lisa, no suele pasar la línea media + dolor abdominal + vómitos + hematuria + hipertensión (60%) por aumento de la actividad de la renina + fiebre e hipercalcemia + poliglobulia
Al ser un tumor retroperitoneal no suele causar obstrucción intestinal. METÁSTASIS: - locales: por invasión de la cápsula renal y por vía linfática a ganglios regionales - distales: hematógenas: pulmonares 85% y hepáticas 15% |
|
ESTADIOS DEL TUMOR DE WILMS
|
1. Resecado + no infiltra
2. Resecado e infiltra cápsula y seno renal 3. Quedan residuos + infiltración peritoneal + ganglios postivos 4. Metástasis hematógenas 5. Tumores renales bilaterales |
|
TRATAMIENTO DEL TUMOR DE WILMS
|
PROTOCOLO SIOP
quimioterapia preoperatorioa reductora + extirpación quirúrgica + QT postoperatoria +/-RT Pronóstico de supervivencia del 90% |
|
RETINOBLASTOMA
|
Es un tumor que se origina en la célula embrionaria de la retina y se debe a defectos en el gen del retinoblastoma (RB1). Es el tumor intraocular más frecuente en pediatría con una incidencia del 3% de las neoplasias infantiles.
|
|
FORMAS CLÍNICAS: ESPORÁDICO Y HEREDITARIO
|
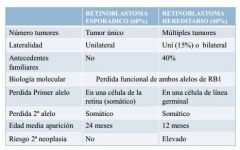
Se manifiesta de diferentes formas:
- leucocoria (60%) - estrabismo (20%) - heterocromia - ojo rojo |
|
DIAGNÓSTICO
|
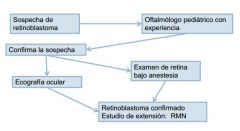
|
|
TRATAMIENTO
|
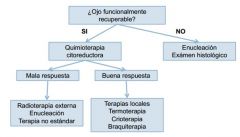
|
|
RABDOMIOSARCOMA
|
Es un tumor maligno embrionario derivad de células mesenquimales cuya diferenciación a células de músculo estriado maduro es incompleta. Puede localizarse en cualquier parte del organismo, incluyendo lugares donde no hay músculo esquelético.
Es el tumor de tejidos blandos más frecuente del niño. Y constituye un 5% del cáncer pediátrico. Se asocia a anomalías congénitas del SNC y genitourinarias. |
|
HISTOLOGÍA
|
Según la histología varía el pronóstico:
- BOTROIDE: buen pronóstico - EMBRIONARIO: pronóstico intermedio - ALVEOLAR: mal pronóstico |
|
CLÍNICA DEL RABDOMIOSARCOMA
|
La clínica depende de su localización: genito-urinaria, cabeza y cuello, extremidades, órbita, otras..
|
|
ESTADIOS
|
-Grupo I:
Resección completa, márgenes negativos -Grupo II: Resección macroscópica, evidencia extensión locoregional -Grupo IIa: Resección completa, márgenes positivos -Grupo IIb: Resección completa, márgenes negativos, ganglios linfáticos positivos -Grupo IIc: Resección completa, márgenes positivos, ganglios linfáticos extirpados positivos -Grupo III: Enfermedad residual macroscópica, incluyendo ganglios linfáticos no resecados -Grupo IV: Metástasis a distancia presentes al diagnóstico |
|
TRATAMIENTO
|
LOCAL: cirugía y o radioterapia
QUIMIOTERAPIA: citorreducción previa al tratamiento local y control de la diseminación a distancia VAC e IVA |