- Barajar
ActivarDesactivar
- Alphabetizar
ActivarDesactivar
- Frente Primero
ActivarDesactivar
- Ambos lados
ActivarDesactivar
- Leer
ActivarDesactivar
Leyendo...
Cómo estudiar sus tarjetas
Teclas de Derecha/Izquierda: Navegar entre tarjetas.tecla derechatecla izquierda
Teclas Arriba/Abajo: Colvea la carta entre frente y dorso.tecla abajotecla arriba
Tecla H: Muestra pista (3er lado).tecla h
Tecla N: Lea el texto en voz.tecla n
![]()
Boton play
![]()
Boton play
![]()
29 Cartas en este set
- Frente
- Atrás
|
Definicion receptor farmacológico
|
Moleculas con que los fármacos son capaces de interactuar selectivamente, generando una modificación constante y específica en la función celular
|
|
Función receptor farmacológico
|
Union de ligando y transmisión de señal: genera una activación o inhibición del receptor
|
|
Criterios de reconocimiento y unión de fco-receptor
|
Reversibilidad, Saturabilidad, Estereoselectividad, Especificidad al agonista y especificidad tisular
|
|
Describe reversibilidad
|
Se relaciona con la fuerza de enlace, la interacción más fuerte es la covalente, le sigue el enlace iónico, puentes de hidrógeno, interacción hidrofóbica y fuerzas de Van der Waals.
Los fármacos comúnmente utilizan las uniones débiles. La variación de energía libre de gibbs para desplazar el equilibrio de la formación fco-receptor de 1% a 99% debe ser aproximadamente -5.5 kcal/mol |
|
Describa la saturabilidad
|
Los receptores están en un número finito, si se ocupan todos tendremos el efecto máximo esperable
|
|
Describa la estereoselectividad
|
Tenemos que verificar si nuestro fármaco posee un carbono quiral el cual le otorgará estereoisomeros con una orientación tridimensional diferente, este puede o no ser relevante para el sitio de unión.
Si es relevante, los estereoisomeros podrían tener diferente efecto y/o constante de disociación (Kd). En caso de que no lo sea, el administrar una mezcla racémica no afectaría la unión del fármaco al receptor |
|
¿A qué nos referimos con transducción?
|
La unión al receptor de un agonista debe ser transducida en alguna respuesta funcional. Una vez que el fármaco se una al receptor, generará un efecto celular, luego un efecto fisiológico y finalmente un efecto clínico.
De los diferentes receptores a los cuales se puede unir el fármaco tenemos los canales iónicos que son los más rápidos (ms), le siguen los acoplados a proteína G (s), unidos a quinasa (h) y receptores nucleares (h). No solo se puede unir a receptores, tambien puede unirse al ADN como los antibióticos, antivirales, anticáncerigeno, etc. |
|
¿Que es el efecto farmacológico?
|
Es una respuesta cuantificable en unidades precisas
|
|
Describir la curva dosis-respuesta
|
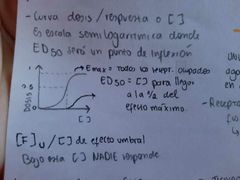
El efecto máximo nos permite normalizar a una escala entre 0-1 o 0-100%
Es importante saber que la diferencia entre un efecto benéfico y tóxico es la DOSIS, ademas de que un mismo fármaco puede tener diferentes efectos y más de uno. En la gráfica curva/respuesta podemos poner el efecto farmacológico, tóxico y letal. |
|
¿Qué es el indice terapéutico?
|

Medida de seguridad para un fármaco
IT= [tóxica]50/[efectiva]50 Mientras más alto sea el índice terapéutico, más seguro será el fármaco. Si tenemos un IT=10, significará que necesitaremos 10 veces la concentración del fármaco para llegar a la toxicidad. |
|
Formacion complejo farmaco-receptor
|
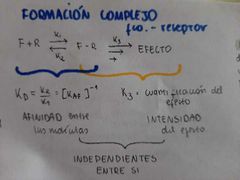
|
|
¿Cuál es la teoría del equilibrio?
|
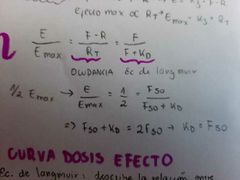
Está la teoría de Clark la cual incluye la fórmula del número total de receptores y la ecuación de langmuir que describe la OCUPANCIA (proporción de receptores ocupados)
También tenemos la teoría de Ariens la cual nos dice que el efecto es proporcional a los receptores ocupados por el fármaco (E=K3 × RT) y que el efecto máximo es proporcional a los receptores totales (Emáx=K3 × RT) Además de que Kd=ED50 |
|
¿Qué relación tiene la ecuación de Langmuir con la curva dosis efecto?
|
Recordemos que la ecuación de langmuir nos describe la relación entre Emáx (ocupancia) y la concentración de fármaco libre.
Como Kd = ED50, podemos decir que con el eje de concentración podría obtener la afinidad del fármaco, ya que en este gráfico se evidencia el ED50, por lo tanto tensremos la Kd y de esta la constante de afinidad (Kd = (1/Kaf)) |
|
Cuantificación del efecto
|
Para una serie homóloga de fármacos (para un mismo receptor o efecto clínico) podemos cuantificar el efecto con K3 a la cual llamaremos actividad intrínseca.
K3 puede tener valor de 1 (o 100%) que se asociará a un agonista completo, es decir, que ejerce un efecto máximo. El valor puede ir bajando hasta llegar a 0 donde no se producirá efecto pero SÍ SE UNE AL RECEPTOR, a esto le llamaremos antagonista competitivo. |
|
¿Qué es la potencia?
|

Está relacionada con la concentración, a menor ED50 más potente será
|
|
¿Qué es la eficacia?
|

Se relaciona con la intensidad del efecto, aquel que tenga 100% o actividad intrínseca 1 será el agonista completo
|
|
Tipos de agonista
|
Agonista completo
Agonista parcial Antagonista neutral Agonista inverso Antagonista competitivo estricto Dualismo: sinergia y antagonismo Antagonismo No competitivo estricto Antagonismo No competitivo |
|
¿Cual es la gráfica de un agonista completo?
|
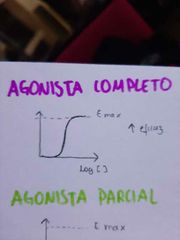
|
|
¿Cuál es la gráfica de un agonista parcial?
|
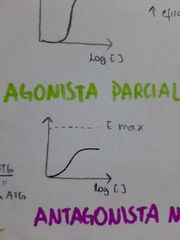
|
|
¿Cuál es la gráfica de un antagonista neutral?
|

Llevan el receptor a su linea base
|
|
¿Cuál es la gráfica y qué es un agonista inverso?
|

Hace que los receptores pasen de activos a inactivos
|
|
Describa un antagonista competitivo estricto
|

Junto con el agonista competiran por el receptor. Mantendrá un efecto máximo, habrá un aumento aparente de la Kd y abrá un cambio de la concentración del efecto umbral.
El agonista deberá aumentar su concentración para desplazar al antagonosta |
|
Describa el dualismo
|

Agonista (A) y agonista parcial (B) compiten por el mismo receptor, en un principio ocurre sinergia (suma de los efectos) debido a que tanto A como B ocupan sus receptores ejerciendo su propia K3 y por consiguiente su propio efecto. Luego B ocupa todos sus receptores y A no producirá su efecto máximo hasta que aumente su concentración y desplace a B
|
|
Describa el antagonismo no competitivo estricto
|

Como es NO COMPETITIVO cada fármaco tendrá su propio receptor y el antagonista tendrá una K3 negativa, de -1 en este caso por ser estricto, por lo que modularía completamente de manera negativa al otro receptor.
En este caso NO SE SOLUCIONA aumentando las concentraciones del agonista ya que ocupan receptores distintos |
|
Describa el antagonismo no competitivo
|
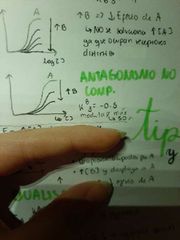
Cada fármaco tendrá su propio receptor y el antagonista tendrá una K3 negativa. Ejemplo K3=-0.5 significa que modula como máximo en un 50% al agonista
|
|
¿Cómo se cuantifica la eficacia y la afinidad?
|
La eficacia se cuantifica con la actividad intrínseca (K3) y la afinidad con la Kd o ED50
|
|
¿Cómo se cuantifica un agonista?
|
Con la fórmula pDx= -log [F] con x=Emáx/E
Usualmente se utiliza en ED50, para calcular el X en esta situación debemos tener en cuenta la siguiente relación: Emax/E = 1/0.5 = 2 por lo tanto, sera pD2= -log[F]50 pero como según la ecuación de langmuir la ED50=KD, tendremos que pD2= -logKD y como la KD= 1/Kaf finalmente obtendremos pD2= logKaf Esto quiere decir que A MAYOR PD2 MAYOR POTENCIA, debido a que tendrá menor KD |
|
¿Cómo se cuantifica un antagonista competitivo estricto?
|
pAx= -log [ATG comp] con x= ([F] en presencia del ATG)/[F] en ausencia del ATG]
|
|
¿Cómo se cuantifica un antagonista no competitivo?
|
pD'x= -log[ATG no comp] con x=Emáx/EmáxATG
|